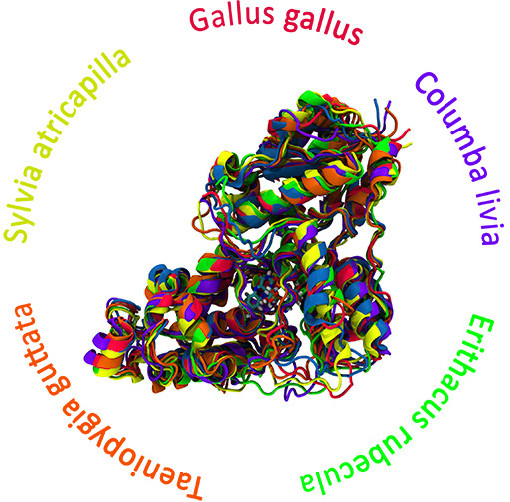
The prediction of protein structures presents a formidable challenge due to the intricate nature of protein folding, a complex puzzle in molecular biology. While achieving accurate protein models is arduous, advancements in computational tools like homology modeling or the revolutionary AlphaFold algorithm have substantially streamlined this process. These computational approaches enable the rapid generation of protein models, marking significant progress in this field.
Our research group is delving into the enigmatic world of cryptochrome proteins—a class of proteins hypothesized to play a pivotal role in the biological phenomenon of magnetoreception in animals. We employ extensive Molecular Dynamics (MD) simulations to reconstruct the atomistic structures of cryptochromes. These simulations facilitate the exploration of biomolecular systems, enabling us to probe the time evolution of cryptochrome proteins over microseconds through all-atomic simulations and even longer timescales using coarse-grained methods.
This project aims to unlock more profound insights into various properties governing cryptochrome behavior through this comprehensive time-evolution study. This includes elucidating activation mechanisms, unraveling ligand binding dynamics, assessing structure prediction stability, and comprehending other dynamic processes occurring on the microsecond timescale. By leveraging computational tools to dissect the intricate dance of atoms within cryptochrome proteins, we strive to unravel the secrets behind their functionality and contribution to animal magnetoreception.