Virtual screening serves as a pivotal computational method within the realm of drug discovery, facilitating the exploration of vast compound libraries in search of those elusive molecular structures with the potential to form crucial interactions with a drug target. In most cases, this target is a protein receptor or enzyme intricately linked to specific diseases or conditions. The ultimate objective is to pinpoint and prioritize the most promising compounds, ones that warrant further labor-intensive experimental assessments. This strategic approach not only streamlines the drug discovery process but also offers the tantalizing prospect of significantly curtailing the associated time, financial resources, and the sheer volume of experiments necessary to identify medically active molecules.
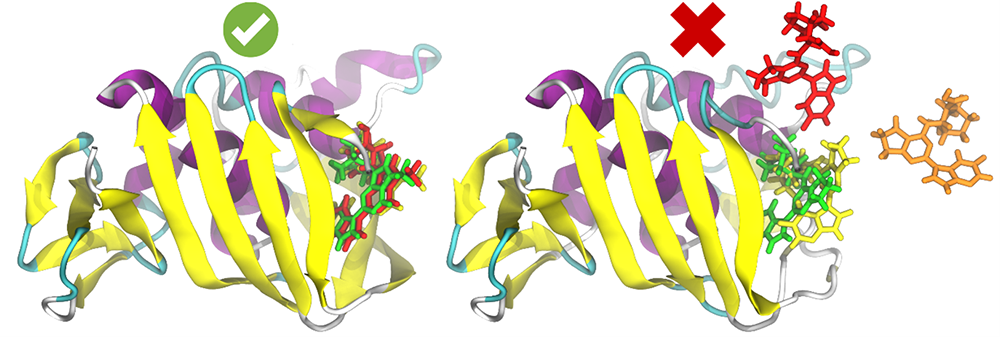
Within our research group, we are at the forefront of drug discovery through the pioneering application of virtual screening. This computational technique allows us to meticulously search vast compound libraries for those elusive structures with a high likelihood of binding to drug targets. These targets, often intricate protein receptors or enzymes intricately linked to various diseases, are the focal points of our research efforts. Our core mission involves identifying the most promising compounds that merit further experimental testing. This approach not only streamlines the drug discovery process but, significantly, dramatically reduces the time, cost, and sheer number of experiments required to unearth medically active molecules.\p>