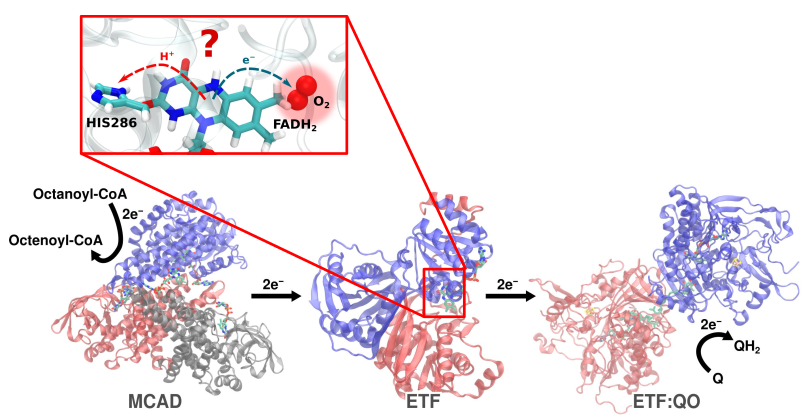
Survival in organisms intricately relies on orchestrating enzymatic processes within the mitochondria, where cellular energy conversion unfolds. This metabolic journey involves a cascade of redox reactions and electron transfers. Simultaneously, molecular oxygen is an indispensable participant in this energy extraction process. However, the cohabitation of these essential components is not without peril. Molecular oxygen has the potential to disrupt electron transfer within enzymes, culminating in the generation of superoxide radicals. These highly reactive species can perturb critical cellular processes involved in energy conversion. Even at low concentrations, superoxides exert cytotoxic effects, threatening cellular viability. Furthermore, the formation of superoxides is implicated in cellular aging.
This intricate dance between molecular oxygen and enzymes, with potential repercussions on cellular aging, is a focal point of our research group. A thorough comprehension of molecular oxygen's binding and unbinding dynamics within enzymes is imperative for a nuanced exploration of cell aging. Identifying potential oxygen-binding sites in enzymes precedes the scrutiny of the spatial distribution of molecular oxygen, a task undertaken through molecular dynamics simulations. These simulations enable the observation of potential electron transfer pathways, elucidating the transformation of molecular oxygen into superoxide radicals. Thus, our research offers insights into the molecular mechanisms underlying cellular aging, contributing to the broader understanding of biological processes and pathologies.